Cell tradition
All cells have been cultured at 5% CO2 and 37 °C. Suspension cell traces together with OCI-AML2, OCI-AML3, Nalm6, MOLM-13, Jurkat, Jeko1, SupT1, K562, HL-60 and Kasumi-1 have been maintained by centrifuging cells, eradicating the medium and resuspending cells in contemporary full medium. Suspension cell cultures have been cut up when cell density reached 2 million cells per ml. Tradition medium used was 1× RPMI 1640 base medium (Thermo Fisher Scientific) with 1% penicillin–streptomycin (Pen/Strep) and 10% heat-inactivated FBS (Thermo Fisher Scientific). OCI-AML2 and OCI-AML3 cells have been cultured in MEM α medium (Fisher Scientific) supplemented with 20% FBS (Fisher Scientific) and 1% Pen/Strep. A549, MC38, A673, SH-SY5Y, H4, U251, MDA-MB-231, B16F10 and first mouse PDAC1-10 (ref. 56) cells have been cultured in DMEM medium (Fisher Scientific) supplemented with 10% FBS (Fisher Scientific) and 1% Pen/Strep. LA-N-5 cells have been cultured in DMEM (Fisher Scientific) supplemented with 20% FBS (Fisher Scientific) and 1% Pen/Strep. KNS-42 cells have been cultured in DMEM (Fisher Scientific) supplemented with 10% FBS (Fisher Scientific), 1% sodium pyruvate and 1% Pen/Strep. UT-SSC-42B cells have been cultured in DMEM (Fisher Scientific) supplemented with 10% FBS (Fisher Scientific), 1% NEAA and 1% Pen/Strep. Calu-1, HCT-116, MHH-ES-1 and U2OS cells have been cultured in McCoy’s 5A medium (Fisher Scientific) supplemented with 10% FBS (Fisher Scientific) and 1% Pen/Strep. SaOS cells have been cultured in McCoy’s 5A (Fisher Scientific) supplemented with 15% FBS (Fisher Scientific) and 1% Pen/Strep. HOS, MG-G3, FADU, DETROIT, HT1080 and HPAF-II cells have been cultured in MEM medium (Fisher Scientific) supplemented with 10% FBS (Fisher Scientific) and 1% Pen/Strep. NCI-H520, KYSE-30, KYSE-140, SiMa, PFSK1, PC3, 22Rv1, ASPC1, BXPC3, SU86.86 and YAPC cells have been cultured in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% FBS (Fisher Scientific) and 1% Pen/Strep. OE21 and OE33 cells have been cultured in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% FBS (Fisher Scientific), 2% l-glutamine (Thermo Fisher Scientific) and 1% Pen/Strep. All the first murine AML cells have been cultured in X-VIVO 20 medium with gentamicin and PR1 (Lonza) supplemented with 5% FBS, recombinant murine IL-3 (10 ng ml−1, PeproTech), IL-6 (10 ng ml−1, PeproTech) and SCF (50 ng ml−1, PeproTech) and 1% penicillin–streptomycin–glutamine (Gibco). Cell traces have been acquired from ATCC and the Sanger Institute Most cancers Cell Assortment except in any other case famous. A full listing of the cell traces’ supply is supplied within the Reporting abstract. Cell cultures have been periodically checked for Mycoplasma and maintained as Mycoplasma unfavorable. Human cell traces employed have been both not listed within the cross-contaminated or misidentified cell line database curated by the Worldwide Cell Line Authentication Committee or have been beforehand verified by karyotyping.
Ex vivo tradition of murine main leukemia
Flt3ITD/+ (Flt3 inner tandem duplication) mice have been kindly supplied by G. Gilliland (Harvard Medical College, USA) and crossed with Rosa26Cas9/+ mice. Freshly remoted BM from 6–10-week-old feminine Flt3ITD/+;Rosa26Cas9/+ or moribund Npm1fl−cA/+;Flt3ITD/+;Rosa26Cas9/+, Npm1fl−cA/+;NrasG12D/+ mice have been used. BM cells have been uncovered to erythrocyte lysis (BD Pharm Lyse, BD Biosciences), adopted by magnetic bead collection of Lin− cells utilizing the Lineage Cell Depletion Equipment (Miltenyi Biotec) in accordance with the producer’s directions. Lin− cells have been cultured in X-VIVO 20 (Lonza) supplemented with 5% FBS (Life Applied sciences), 10 ng ml−1 IL-3 (PeproTech), 10 ng ml−1 IL-6 (PeproTech) and 50 ng ml−1 SCF (PeproTech) and 1% penicillin–streptomycin–glutamine. The retroviral constructs pMSCV-MLL-AF9-IRES-YFP and pMSCV-MLL-ENL-IRES-Neo have been used with the bundle plasmid psi-Eco to provide retrovirus. Subsequent, 293T cells (Life Applied sciences) have been cultured and ready for transduction in 10-cm plates. For virus manufacturing, 5 μg of the above plasmids and 5 μg of the psi-Eco packaging vector have been transfected dropwise into the 293T cells utilizing 47.5 μl TransIT-LT1 (Mirus) and 600 μl Opti-MEM (Invitrogen). The ensuing viral supernatant was collected as beforehand described. Transduction of main Flt3ITD/+;Rosa26Cas9/+ mouse cells was carried out in six-well plates as talked about above. After transduction, transduced cells have been sorted for YFP (for MLL-AF9) or chosen with neomycin (for MLL-ENL).
Dissection and tradition of main mouse prostate tissue
Regular prostate tissue was derived from Rosa26-LSL-Cas9 knockin mice on B6J (pressure 26175, Jackson Laboratory), and Pten−/−;Trp53−/− prostate tumors have been derived from a mouse mannequin of prostate most cancers of the identical genetic background just like Feng et al.57. Tissues have been minced into small items and transferred right into a gentleMACS C tube (Miltenyi Biotec) for enzymatic digestion utilizing the Multi Tissue Dissociation Equipment 1 (130-110-201, Miltenyi Biotec). Digestion was carried out in a gentleMACS Dissociator utilizing this system 37C_Multi_A. Following enzymatic dissociation, the cell suspension was pelleted, resuspended and filtered by means of a 70‐μm cell strainer. Regular and tumoral dissociated prostate cells have been cultured as 3D organoids utilizing the protocol and medium composition described by Drost et al.58. Cultures have been maintained below these situations for six–7 days to complement for epithelial cells, after which cells have been maintained as monolayer cultures on plates coated with collagen I (354236, Corning) utilizing the identical medium composition.
Lentiviral vector manufacturing and an infection
For virus manufacturing, 293FT cells have been transfected with the lentiviral vector (lentiCRISPR-v2) both as an empty vector or containing NPM1 information RNA along with the packaging plasmids psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259). The viral supernatant was collected 48 and 72 h after transfection and concentrated in a single day at 6,000g and 4 °C. A complete of 1 × 106 cells and viral supernatant have been blended in 2 ml tradition medium supplemented with 8 μg ml−1 polybrene (Millipore), adopted by spinfection (60 min, 900g, 32 °C), and have been additional incubated in a single day at 37 °C. The medium was refreshed on the next day, and the transduced cells have been cultured additional. Pellets for protein have been collected 7 and 9 days after transduction.
Protein extraction and western blot for the knockout experiment
OCI-AML2 cells have been transduced with both NPM1 gRNAs or management gRNAs. Cell pellets have been collected on day 7 after transduction and lysed with whole-cell lysis buffer (0.2% Nonidet P-40, 50 mM Tris-HCl, pH 8.0, 450 mM NaCl, 1 mM EDTA), 1× protease inhibitor cocktail 1 (Merck), 1× phosphatase inhibitor 2 (Merck), 1× phosphatase inhibitor 3 (Merck) and 1 mM DTT (Epigentek)) and incubated on ice for 10 min. The lysates have been then centrifuged at 20,000g and 4 °C for 10 min. The supernatant was transferred to a contemporary tube and quantified utilizing the Bradford assay (Bio-Rad). Following quantification, the samples have been supplemented with 1× LDS pattern buffer (Thermo Fisher Scientific) and 1× Pattern Lowering Agent (Thermo Fisher Scientific), and so they have been then incubated at 70 °C for 10 min. Subsequent, 10 μg protein was loaded. Western blotting was carried out utilizing SDS–PAGE gels, and samples have been blotted onto a PVDF membrane. It was carried out utilizing the next antibodies: anti-NPM1 (FC8791), anti-H3 (Abcam, ab1220) as a loading management at a 1:1,000 dilution and goat anti-mouse IgG H&L, HRP conjugated (Abcam, ab205719) at a 1:10,000 dilution.
Blood counts
For blood counts, 20 μl blood was collected from the tail vein of mice utilizing a capillary pipette containing anticoagulants (EDTA). The EDTA anticoagulated blood samples have been used to acquire a whole blood depend with a Vet ABC analyzer (Horiba ABX). Samples have been counted not than 20 min after blood was drawn.
Actual-time PCR
For Fig. 4a, genomic DNA was extracted from murine PB utilizing the DNeasy Blood and Tissue Equipment (Qiagen). Genomic DNA (10 ng) was used, and the degrees of Cas9 and Gapdh have been analyzed on a QuantStudio 5 real-time PCR instrument (Utilized Biosystems) utilizing PowerUp SYBR Inexperienced Grasp Combine (Utilized Biosciences). The relative quantification of Cas9 was carried out utilizing the comparative cycle threshold (Ct) technique in opposition to the housekeeping gene Gapdh. The primer sequences are listed in Supplementary Desk 3.
PCR
For Prolonged Information Fig. 6b, genomic DNA was extracted from murine PB 2 weeks after injection utilizing the DNeasy Blood and Tissue Equipment (Qiagen). Primers for Flt3ITD have been used for PCR amplification of genomic DNA (20 ng). The PCR product was analyzed by agarose gel electrophoresis, stained with GelRed. The 1 kb plus DNA Ladder (NEB) was used.
Antibody staining and move cytometry evaluation of AML cells
For main murine AML experiments associated to Fig. 2b and Prolonged Information Figs. 2b and 3b, 6–10-week-old C57BL/6 male mice have been injected with 106 main murine AML cells by intravenous injection from the indicated animal AML fashions, as described above. Upon animal illness, BM was remoted and lysed in 0.85% NH4Cl for five min. Main antibodies, at a focus of 0.5 μg per response, both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell), have been precomplexed with a 1:1,000 dilution of the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) for 30 min. For intracellular staining, cells have been first permeabilized with 0.1% Triton X-100 (Sigma) for 10 min at room temperature and rinsed with 2% FBS in 1× PBS. For cell floor staining (‘dwell cell’), cells have been processed immediately. BM cells have been blocked in 2% FBS in 1× PBS for 30 min on ice after which stained with the precomplexed mixture of antibodies as acknowledged above. Cells have been washed as soon as with 150 µl of two% FBS in 1× PBS and resuspended in 2% FBS in 1× PBS containing 0.1 µg ml−1 DAPI (Sigma). For intracellular staining, cells have been mounted with 4% formaldehyde adopted by a wash with 2% FBS in 1× PBS. Circulation cytometry evaluation was carried out utilizing a CytoFLEX instrument (Beckman Coulter) and analyzed utilizing FlowJo (model 10, BD).
For main murine AML experiments associated to Fig. 4d, 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy after which injected with 106 main murine AML cells by intravenous injection from the indicated animal AML mannequin, as described above. On day 15 after transplantation, mice have been handled i.p. with a single dose of both 5 mg kg−1 of the mouse IgG2a isotype management antibody or 5 mg kg−1 of anti-NPM1 (mAb2) antibody. On day 18 after transplantation, PB was remoted and lysed in 0.85% NH4Cl for five min. Circulation cytometric evaluation of YFP+ cells was carried out as above.
For Fig. 2c, K562 cells have been transduced with the lentiviral cDNA constructs pKLV-TY1-NPM1-PURO or pKLV-TY1-NPM1c-PURO (mutant NPM1) or an empty pKLV-TY1-PURO vector management. Transduced cells have been chosen with puromycin, after which dwell cells have been stained with both a mouse anti-B23 NPM1 antibody (Merck) or a mouse anti-Ty1 antibody (Diagenode) for 45 min adopted by a staining with a 1:1,000 dilution of the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) for 45 min. Cells have been washed as soon as with 2% FBS in 1× PBS and at last resuspended in 2% FBS in 1× PBS containing 0.1 µg ml−1 DAPI. Circulation cytometry evaluation was carried out as above.
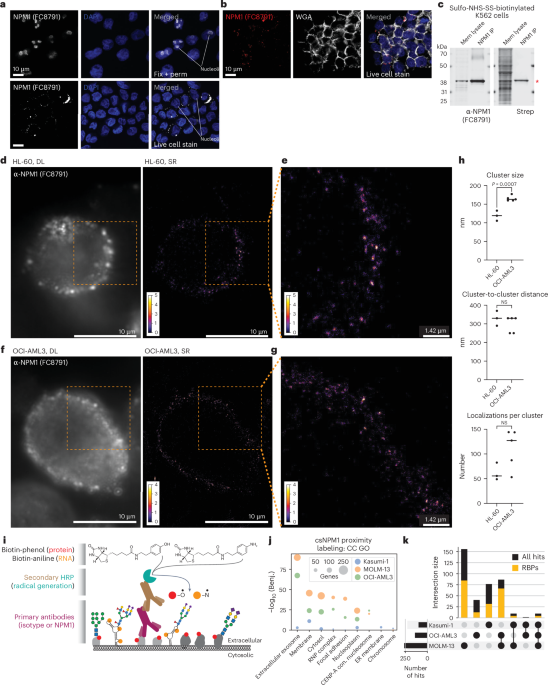
For Prolonged Information Fig. 2g, K562 and OCI-AML3 cells have been incubated with human Fc block (BioLegend, 101319). Afterward, they have been incubated with WGA conjugated to fluorescein (Vector Laboratories, FL-1021-5) at a focus of 1:1,000. When colabeled with FC8791 or mAb2, antibodies have been precomplexed with anti-mouse IgG Alexa Fluor 647 at a ratio of two:1 earlier than being incubated with cells. The ultimate focus of FC8791 and mAb2 was 5 µg ml−1, and the secondary antibody was used at 2.5 µg ml−1. Cells have been washed after binding, subsequently stained with DAPI after which utilized to slides utilizing a Cytospin. Photographs have been obtained on a Leica TCS SP8 microscope.
For AML PDX experiments associated to Prolonged Information Fig. 6e,f, 6–10-week-old SCID-CB17 feminine mice (Charles River, pressure 236) have been injected with 106 patient-derived AML cells by intravenous injection. BM was remoted and lysed in 0.85% NH4Cl for five min. Main antibodies, at a focus of 0.5 μg per response, both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell), have been precomplexed with a 1:1,000 dilution of the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) for 30 min. BM cells have been blocked in 2% FBS in 1× PBS for 30 min on ice after which stained with the precomplexed mixture of antibodies as acknowledged above. Cells have been washed as soon as with 150 µl of two% FBS in 1× PBS and resuspended in 2% FBS in 1× PBS containing 0.1 µg ml−1 DAPI. Circulation cytometry evaluation was carried out as above.
For main murine experiments associated to Fig. 5d, freshly remoted BM from male 20-week-old WT and preleukemic Npm1fl−cA/+ or moribund Npm1fl−cA/+;Flt3ITD/+ mice was used. BM cells have been uncovered to erythrocyte lysis (BD Pharm Lyse, BD Biosciences), adopted by magnetic bead collection of Lin− cells utilizing the Lineage Cell Depletion Equipment (Miltenyi Biotec) in accordance with the producer’s directions. Main antibodies, at a focus of 0.5 μg per response, both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell), have been precomplexed with a 1:1,000 dilution of the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) for 30 min. BM cells have been blocked in 2% FBS in 1× PBS for 30 min on ice after which stained with the precomplexed mixture of antibodies as acknowledged above. Cells have been washed as soon as with 150 µl of two% FBS in 1× PBS and resuspended in 2% FBS in 1× PBS containing 0.1 µg ml−1 DAPI. Circulation cytometry evaluation was carried out as above.
For main murine AML experiments associated to Fig. 5a,b, 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy after which injected with 106 main murine AML cells by intravenous injection from the indicated animal AML mannequin, as described above. Upon animal illness on day 18 after transplantation, BM was remoted and lysed in 0.85% NH4Cl for five min. BM cells have been resuspended in 10% DMSO in FBS and saved at −80 °C for additional functions. BM cells have been thawed and suspended in PBS supplemented with 2% FBS and stained with biotin anti-mouse Ly-6A/E (SCA1) (BioLegend), biotin anti-mouse CD127 (BioLegend, 135005), biotin anti-mouse CD3 (BioLegend, 100201), biotin anti-mouse TER-119/erythroid cells (BioLegend, 116203), biotin anti-mouse/human CD45R/B220 (BioLegend, 103203), BV605 anti-mouse GR1 (BioLegend, 108439), BV650 anti-mouse CD11b (BioLegend, 101239), PerCP/Cy5.5 anti-mouse CD16/CD32 (BioLegend, 101323), PE anti-mouse CD93 (BioLegend, 136503), APC anti-mouse CD48 (BioLegend, 103411), APC/Fireplace 750 anti-mouse CD117 (c-Equipment) (BioLegend, 135139), Zombie aqua viability dye (BioLegend, 423101) and BV421 streptavidin (BioLegend, 405226). All of the antibodies have been used at a 1:400 dilution, other than the viability dye, which was used at a 1:1,000 dilution. The samples have been then stained with both anti-NPM1 (mAb2) antibody or the IgG2a isotype management (Bio X Cell, BE0085), precomplexed with anti-mouse IgG Alexa Fluor 594 (Abcam, ab150108) as a secondary antibody. FMO controls have been included within the experiments to offer a measure of spillover in every channel. This enables for proper gating in every experimental pattern. Circulation cytometry evaluation was carried out utilizing the Cytek Aurora spectral analyzer and analyzed utilizing FlowJo (model 10, BD). Information on this part have been plotted utilizing GraphPad Prism (model 9).
Antibody staining and move cytometry evaluation of stable most cancers fashions
Main antibodies, at a focus of 0.5 μg per response, both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell), have been precomplexed with a 1:1,000 dilution of the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) for 30 min. A complete of 5 × 104 cells for every mannequin utilized in Fig. 6 (Fig. 6a,b,e,h) have been blocked in 2% FBS in 1× PBS for 30 min on ice after which stained with the precomplexed mixture of antibodies as acknowledged above. Cells have been washed as soon as with 150 µl of two% FBS in 1× PBS and resuspended in 2% FBS in 1× PBS containing 0.1 µg ml−1 DAPI (Sigma). Circulation cytometry evaluation was carried out utilizing the CytoFLEX instrument (Beckman Coulter) and analyzed utilizing FlowJo (model 10, BD).
In vivo therapy of regular, main murine AML and PDX fashions
For experiments associated to Prolonged Information Fig. 4d,e, 16–20-week-old C57BL/6 male mice have been used, which have been housed at Boston Kids’s Hospital. All mouse procedures and protocols have been accepted by the Animal Care and Use Committee of Boston Kids’s Hospital and adopted all related pointers and rules. After euthanasia, PB, liver, spleen and BM have been harvested.
For experiments associated to Prolonged Information Fig. 5a,b, 6–10-week-old C57BL/6 male mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of 4 weeks (whole of 4 remedies). Weights have been recorded, and PB from the tail vein was collected on the indicated time factors. Weight measurements of the indicated mouse organs have been taken from all handled cohorts on the finish of the research, on the twenty eighth day after initiation of the related remedies.
For experiments associated to Fig. 4d–f and Prolonged Information Fig. 5c,d, 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy. On day 12 after irradiation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of 4 weeks (whole of 4 remedies). Weights have been recorded, and PB from the tail vein was collected on the indicated time factors. Weight measurements of the indicated mouse organs have been taken from all handled cohorts on the finish of the research, on the 66th day after irradiation.
For main murine Npm1c;Flt3ITD/+/Cas9 AML experiments associated to Fig. 4a–c and Prolonged Information Fig. 6a, 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy after which injected with 106 main murine AML cells by intravenous injection from the indicated animal AML mannequin, as described above. On day 14 after transplantation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of 4 weeks (whole 4 of remedies). Weight measurements of leukemic spleens have been taken from every animal after humane endpoints have been reached.
For main murine MLL-AF9/Flt3ITD/+/Cas9 AML experiments associated to Fig. 4g,h, 6–10-week-old NSG male mice have been injected with 106 main murine AML cells by intravenous injection from the indicated animal AML mannequin, as described above. On day 14 after transplantation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of three weeks (whole of three remedies). Weight measurements of leukemic spleens have been taken from every animal after humane endpoints have been reached.
For xenotransplantation AML experiments associated to Fig. 4i, 6–10-week-old SCID-CB17 (Charles River, pressure 236) feminine mice have been injected with 2 × 106 OCI-AML3 human AML cells by intravenous injection. On day 14 after transplantation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of three weeks (whole of three remedies).
For AML PDX experiments associated to Fig. 4j,ok, 6–10-week-old SCID-CB17 (Charles River, pressure 236) feminine mice have been injected with 106 patient-derived AML cells by intravenous injection. On day 14 after transplantation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses, as soon as per week for a complete of three weeks (whole of three remedies). PB from the tail vein was collected on day 24 after transplantation.
For main murine MLL-AF9/Flt3ITD/+/Cas9 AML experiments associated to Fig. 5c, 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy after which injected with 106 main murine AML cells from main recipients. For the first recipients (associated to Fig. 4d,e and Prolonged Information Fig. 6c,d), 6–10-week-old C57BL/6 male mice have been sublethally irradiated with a whole-body dose of 5.5 Gy after which injected with 106 main murine AML cells by intravenous injection from the indicated animal AML mannequin, as described above. On day 15 after transplantation, mice have been handled i.p. with a single dose of both 5 mg kg−1 of mouse IgG2a isotype management antibody or 5 mg kg−1 of anti-NPM1 (mAb2) antibody. On day 18 after transplantation, BM was remoted and processed accordingly for secondary transplantation as indicated above. Furthermore, on day 18 after transplantation, PB from the tail vein in addition to weight measurements of spleens, lungs and livers have been taken from every animal after humane endpoints have been reached.
All mice used within the research have been housed in particular pathogen-free situations within the UBS animal amenities of the College of Cambridge. All cages have been on a 12–12-h mild–darkish cycle (lights on, 07:30) in a temperature-controlled and humidity-controlled room. Room temperature was maintained at 72 ± 2 °F (22.2 ± 1.1 °C), and room humidity was maintained at 30–70%. The animals have been culled when leukemia-associated signs occurred or humane endpoints have been reached. All animal research have been carried out in accordance with the Animals (Scientific Procedures) Act 1986, UK and accepted by the Ethics Committee on the College of Cambridge. Randomization and blinding weren’t utilized. All knowledge on this part have been plotted utilizing GraphPad Prism (model 9).
In vivo therapy of mouse stable tumor fashions
For main prostate carcinoma experiments associated to Fig. 6c,d, 8–10-week-old C57BL/6 male mice have been injected with 106 main murine prostate carcinoma cells by subcutaneous injection (1:1 ratio of Matrigel and most cancers cells) from the indicated animal mannequin, as described above. For colorectal carcinoma experiments associated to Fig. 6f,g, 8–10-week-old C57BL/6 male mice have been injected with 5 × 105 MC38 cells by subcutaneous injection (1:1 ratio of Matrigel and most cancers cells). For melanoma experiments associated to Fig. 6i, 8–10-week-old C57BL/6 male mice have been injected with 5 × 105 B16F10 cells by subcutaneous injection (1:1 ratio of Matrigel and most cancers cells). On days 5, 7 and 9 after transplantation, mice got i.p. injections of both anti-NPM1 (mAb2) or the IgG2a isotype management (Bio X Cell, BE0085) antibody on the indicated doses (whole of three remedies). Tumors have been dissected when sizes have been approaching or reached the humane finish level restrict (1.2 cm2) as per animal license: on day 17 after transplantation for prostate tumors, on day 13 after transplantation for colorectal carcinoma tumors and on day 11 after transplantation for melanoma. All tumor sizes have been measured utilizing a digital caliper (Jodsen). All stable tumor fashions used within the research have been housed in particular pathogen-free situations within the UBS animal amenities of the College of Cambridge,. All cages have been on a 12–12-hour mild–darkish cycle (lights on, 07:30) in a temperature-controlled and humidity-controlled room. Room temperature was maintained at 72 ± 2 °F (22.2 ± 1.1 °C), and room humidity was maintained at 30–70%. The animals have been culled when humane endpoints have been reached. All animal research have been carried out in accordance with the Animals (Scientific Procedures) Act 1986, UK and accepted by the Ethics Committee on the College of Cambridge. Randomization and blinding weren’t utilized. All knowledge on this part have been plotted utilizing GraphPad Prism (model 9).
Cell floor biotinylation, immunoprecipitation and western blotting
K562 cells have been cultured as above. Cell floor protein labeling was completed utilizing sulfo-NHS-SS-biotin (APExBIO) as described above, after which crude membrane fractions have been remoted. To acquire cytosolic and membrane fractions24, suspension cells have been immediately resuspended in membrane isolation buffer (10 mM HEPES (Thermo Fisher Scientific), 250 mM sucrose (Sigma), 1 mM EDTA) at 5 million cells per 1 ml. Adherent cells have been collected off the plate by scraping in ice-cold PBS, pelleted after which equally resuspended at 5 million cells per 1 ml of membrane isolation buffer. Cells have been rested on ice for as much as 5 min, moved to a glass Dounce homogenizer (Sigma) after which homogenized utilizing 40–80 strokes to acquire a resuspension of roughly 50% launched nuclei. Overdouncing could cause nuclear rupture and contamination of the cytosolic fraction. After douncing, unbroken cells and nuclei have been pelleted by centrifuging at 4 °C for 10 min at 700g. Supernatants (cytosol and membranes) have been rigorously transferred to a brand new tube, and pellets have been discarded. The supernatants have been once more centrifuged at 4 °C for 30 min at 10,000g. Most (90%) of the supernatant was eliminated and saved as cytosolic fractions. The remaining supernatant was discarded, because it was close to to the membrane pellet. The membrane pellet was gently washed with 500 µl of ice-cold 1× PBS (the pellet was not resuspended right here), the tube was centrifuged briefly, and all supernatant was discarded to depart a cleaned membrane pellet. Lastly, the membrane pellet was resuspended in 500 µl CLIP lysis buffer (50 mM Tris-HCl, pH 7.5, 200 mM NaCl (Sigma), 1 mM EDTA, 10% glycerol (Thermo Fisher Scientific), 0.1% NP-40, 0.2% Triton X-100, 0.5% N-lauroylsarcosine). Each the cytosolic and membrane lysates have been saved at −80 °C for later processing. After isolation and solubilization with CLIP lysis buffer, whole protein quantification occurred utilizing the BCA assay. For every pattern, 5 μg protein was used for enter, and 20 μg was used for both anti-NPM1 (FC8791)-coated bead protein A Dynabeads (Thermo Fisher Scientific) or streptavidin-coated bead (MyOne C1 beads, Thermo Fisher Scientific) enrichments. In each circumstances, 10 μl bead surrey was added to the membrane lysates in 100 μl of CLIP lysis buffer, and binding occurred at 4 °C for 16 h. After binding, the beads have been washed 3 times with high-stringency buffer after which twice with 1× PBS. Proteins have been launched from the beads by heating at 85 °C for 10 min in 20 µl 1× LDS (Thermo Fisher Scientific) and 1 mM free biotin (Thermo Fisher Scientific). Enter and enriched proteins have been analyzed by Western blot, staining with anti-NPM1 antibody (Santa Cruz Biotechnology, sc-32256, FC8791) and streptavidin IR800 (LI-COR Biotechnology) and at last scanning on a LI-COR Odyssey CLx scanner. Fractionation as described above was carried out with out cell floor biotinylation for knowledge in Fig. 1a, and western blots have been creating utilizing anti-NPM1 (Santa Cruz Biotechnology, sc-32256, FC8791), anti-NPM1c (Thermo Fisher Scientific, 32-5200), anti-β-actin (Santa Cruz Biotechnology, sc-47778) and anti-RPN1 (Santa Cruz Biotechnology, sc-48367) antibodies.
mAb2 design and synthesis
VH and VL sequences have been grafted to the fixed area of the mouse IgG2a heavy chain and the mouse λ mild chain, respectively, to generate a brand new mouse antibody (mAb). These sequences got to Curia International. At Curia, the gene synthesis course of entails overlapping oligonucleotide synthesis and meeting, adopted by cloning into Curia’s proprietary high-expression mammalian vector. Manufacturing and mAb high quality management have been carried out by Curia to provide a protein A-purified IgG fraction with low endotoxin (<1 EU per mg IgG) in 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4.
Dwell and glued cell staining (human samples), move cytometry and knowledge evaluation
Cells have been cultured as described above and immediately counted. Sometimes, 50,000 cells have been used and blocked with Human TruStain FcX (Fc block, BioLegend) or Mouse TruStain FcX (Fc block, BioLegend) in move cytometry buffer (0.5% BSA (Sigma) in 1× PBS) for no less than 15 min on ice; cells have been saved on ice from this level ahead. For intracellular staining (‘repair–perm’), cells have been first mounted with 3.7% formaldehyde for 10 min at 25 °C, rinsed as soon as with 1× PBS after which permeabilized with 0.1% Triton X-100 (Sigma) for 10 min at 25 °C and at last rinsed as soon as with 1× PBS. For cell floor staining (‘dwell cell’), cells have been processed immediately after dwell Fc blocking on ice. Precomplexed antibodies have been added to dwell or mounted cells. To precomplex, main unconjugated antibodies together with mouse isotype (Santa Cruz Biotechnology, sc-2025) and anti-NPM1 (Santa Cruz Biotechnology, sc-32256, FC8791) and anti-Ty1 tag (Diagenode) have been certain in answer (precomplexed) to a goat anti-mouse AF647 secondary antibody (Thermo Fisher Scientific, A32728) or a goat anti-mouse AF488 secondary antibody (Thermo Fisher Scientific, A28175) for no less than 30 min on ice earlier than use. The molar ratio was 2:1 (main:secondary). To the blocked cells, precomplexed antibody was added at a remaining focus of 1 µg ml−1 (main antibody) and allowed to bind to cells for 60 min on ice. After staining, cells have been centrifuged at 4 °C for 3 min at 400g, and the supernatant was discarded. All cell centrifugation steps befell utilizing these situations. Cells have been washed as soon as with 150 µl move cytometry buffer, centrifuged below the identical situations and at last resuspended in move cytometry buffer containing 0.1 µg ml−1 DAPI. Information assortment occurred on a BD Biosciences LSRFortessa 3, and a gating technique was used to isolate dwell single cells to look at antibody binding utilizing FlowJo software program (model 10, BD).
The frozen PB and BM samples obtained from wholesome donors in addition to sufferers with AML have been processed with care to make sure as little cell lysis and excessive viability after thawing. Vials have been warmed in a 37 °C water tub for two–3 min after which utterly thawed in 5 ml of ice-cold move cytometry buffer. Cells have been pelleted from this preliminary resuspension at 400g for five min at 4 °C. The supernatant was discarded, and cells have been resuspended in 1 ml of contemporary ice-cold move cytometry buffer and counted. For every pattern, 1 million cells per ml have been taken for Fc blocking and marking in accordance with the above protocol. Subsequent, we stained the cells utilizing the dwell protocol with the precomplexed isotype or anti-NPM1 (mAb2) antibodies for 30 min on ice in 100 µl move cytometry buffer. After that, we centrifuged the cells and discarded the supernatant as earlier than. The cells have been then stained once more with 200 ng of dye-conjugated cell type-specific antibodies (anti-human CD45 (HI30), 304024; anti-human CD34 (581), 343516; anti-human CD117 (104D2), 313204; anti-human CD33 (WM53), 303416; anti-human CD13 (WM15), 301710; anti-human HLA-DR (L243), 307604; anti-human CD3 (OKT3), 317308; and/or anti-human CD19 (HIB19), 302206; all BioLegend) on ice for 30 min in 100 µl move cytometry buffer. Lastly, the cells have been pelleted once more, the supernatant was discarded, one 200-µl move cytometry buffer wash was carried out, and the cells have been lastly resuspended in 200 µl move cytometry buffer with 0.1 µg ml−1 DAPI for move cytometry evaluation. Wholesome donor and AML affected person samples have been obtained with knowledgeable consent below (1) UK moral approval (IRAS reference 340167, beforehand 149581, REC 07-MRE05-44). Moreover, wholesome donor and AML affected person samples have been collected from sufferers situated on the Dana-Farber Most cancers Institute (DFCI) or Brigham and Girls’s Hospital (USA). Samples have been then processed and banked within the Pasquarello Tissue Financial institution in accordance with IRB-22-160 on the DFCI; we obtained help from the Hematologic Malignancies Information Repository to establish affected person samples from the tissue financial institution that have been bona fide AML. The HMDR additionally supplied related info relating to illness traits, reminiscent of cytogenetics, mutations and immunophenotype. Samples have been banked in accordance with DFCI Protocol 01-206: tissue and knowledge assortment for analysis research in sufferers with hematologic malignancies, BM problems and regular donors. Pattern traits have been obtained utilizing DFCI IRB Protocol 22-160. This can be a DFCI-specific tissue-banking protocol, which isn’t obtainable publicly for overview, however it may be shared upon request.
Confocal microscopy pattern preparation, knowledge acquisition and evaluation
For suspension cells, culturing, counting and Fc blocking have been carried out as described above. Samples for ‘dwell cell’ imaging have been processed in accordance with the dwell cell move cytometry protocol famous above; nonetheless, after staining and washing, cells have been mounted with 3.7% formaldehyde (37% inventory, Sigma) for 30 min at 25 °C. Main and secondary antibodies famous above have been used however sequentially, quite than precomplexed: main antibody was added at a remaining focus of two.5 µg ml−1 for 45 min on ice in move cytometry buffer. After staining, cells have been washed twice with 1× PBS after which stained with a secondary antibody at a remaining focus of two.5 µg ml−1. Secondary stains occurred for 30 min on ice and at midnight, after which cells have been washed as soon as with 1× PBS. A remaining fixation for ‘repair–perm’ samples was carried out in parallel with the ‘dwell cell’ samples with 3.7% formaldehyde in 1× PBS for 30 min at 25 °C at midnight. Nuclei have been stained with 0.1 µg ml−1 DAPI in move cytometry buffer for 15 min at 25 °C. Suspension cells have been utilized to glass slides utilizing a Cytospin centrifuge (Fisher Scientific): this was completed by centrifugation at 500g for five min in a Cytospin 1867. Lastly, cells have been mounted in ProLong Diamond Antifade Mountant (Thermo Fisher Scientific), and a coverglass was sealed over the pattern with nail polish. All samples have been then imaged on a Leica TCS SP8 STED ONE microscope. For all experiments, no less than three areas of curiosity have been acquired utilizing a ×63 oil-immersion goal throughout a number of z slices. Leica’s line-sequential scanning technique was used, and pictures have been acquired at 1,024-by-1,024 decision with a pinhole dimension of 1 AU. The DAPI channel was acquired with a PMT detector, whereas all different channels have been imaged utilizing hybrid detectors. For Prolonged Information Fig. 2i, the DAPI channel was imaged utilizing a hybrid detector, and pictures have been analyzed utilizing ImageJ (model 1.54f). Subsequent, utilizing Imaris Microscopy Picture Evaluation software program (Oxford Devices), single slices of the confocal stack have been analyzed.
Tremendous-resolution imaging and reconstruction
Cells have been ready as described above for dwell cell imaging. Right here, main conjugated antibodies (AF647) have been used immediately for cell labeling and included mouse isotype (Santa Cruz Biotechnology, sc-24636-AF647) and anti-NPM1 (Santa Cruz Biotechnology, sc-32256-AF647, FC8791). To keep away from cell motion in the course of the SR acquisition, cells have been immobilized on a glass-bottom plate precoated with poly-l-lysine (Sigma, P4707) and Cell-Tak (Corning, 354240). An summary of the strategy and processing may be present in Prolonged Information Fig. 3a. For single-molecule SR microscopy, we used direct stochastic optical reconstruction microscopy. To carry out this, the PBS wherein the cells have been saved was changed by a lowering oxygen scavenging buffer to induce blinking of fluorophores as described within the literature59. The blinking buffer consisted of two μl ml−1 catalase (Sigma), 10% (wt/vol) glucose (BD Biosciences), 100 mM Tris-HCl (Thermo Fisher Scientific), 560 μg ml−1 glucose oxidase (Sigma) and 20 mM cysteamine (Sigma). First, diffraction-limited photos have been obtained with low-intensity illumination of few W cm−2. Subsequent, the laser energy was elevated to roughly 3 kW cm−2. Picture acquisition was began after a brief delay to make sure that most fluorophores have been shelved right into a darkish state. The publicity time was 50 ms, and roughly 40,000 frames have been recorded.
As the information have been obtained with an sCMOS digicam, which usually exhibit few pixels with deviating sensitivity (‘scorching’ and ‘chilly’ pixels), the obtained single-molecule knowledge have been corrected for particular person pixels with abnormally excessive or low sensitivity first60. In whole, 4,000 frames of a uncooked knowledge stack have been averaged. Cold and warm pixels, that are a scientific deviation, persist, in distinction to the random single-molecule alerts. Every pixel was in comparison with its neighbors utilizing 8-connectivity. If a deviation of greater than 3% from the median of the neighboring pixel was noticed, a correction issue that set the pixel to the median of its neighbors was recorded as beforehand described61. In any other case, the pixel was not thought-about to be considerably brighter or darker. This yielded a correction masks, which was utilized to all frames of the uncooked knowledge. Lastly, the no-light counts have been subtracted from the pixel-corrected knowledge.
For localization of single molecules, the Fiji plugin ThunderSTORM was used62. Every digicam body was filtered with a B-spline filter of order 3 and scale 2. Native maxima, akin to single-molecule alerts, have been detected with eight-neighborhood connectivity and a threshold of 1.1 or 1.2 occasions the usual deviation of the primary wavelet degree. Detected native maxima have been fitted with a 2D Gaussian utilizing least squares, and the place was recorded. Subsequent, to account for single-molecule alerts being energetic in a number of frames, merging of localizations was carried out, utilizing a most distance of 30 nm and a most of 5 off frames with no restrict relating to on frames. Cross-correlation-based drift correction (magnification, 5; bin dimension, 5) was carried out, adopted by filtering of localizations (sigma of the purpose unfold operate between 60 and 270 nm, depth under 37,800 photons, localization uncertainty smaller than 30 nm). For visualization, the ultimate localizations have been reconstructed as 2D histograms with a magnification of 5 (akin to a pixel dimension of 17.7 nm).
For cluster evaluation, an automatic pipeline was established utilizing the uncooked listing of localizations. First, Ripley’s H operate was calculated on three areas with numerous well-separated clusters. The ensuing preferential cluster dimension from the three areas (which was, notably, all the time very related) was averaged, multiplied by a correction issue of 0.45 and used because the seed radius for the next DBSCAN evaluation. This DBSCAN script yielded all particular person clusters, the full variety of clusters, the common variety of factors per cluster and the spatial relation between clusters. The recognized remaining clusters have been then analyzed with respect to their spatial relation, dimension and variety of localizations. This unbiased strategy follows beneficial evaluation procedures not too long ago described63. Crucially, every dataset was subjected to equivalent postprocessing and cluster evaluation steps with no guide intervention, thus avoiding any biases arising from completely different parameter settings. Customized scripts used for this evaluation may be shared upon request.
Cell floor proximity labeling of proteins, peptide technology and mass spectrometry knowledge evaluation
Samples have been ready equally to the move cytometry workflow as described above. Nonetheless, quite than dye-conjugated secondaries, right here an HRP-conjugated secondary (Thermo Fisher Scientific, 31430) was used. The isotype (management) or anti-NPM1 (FC8791) (goal) main unconjugated antibody was precomplexed with an acceptable secondary HRP antibody at 2:1 (main:secondary) for no less than 30 min on ice. Cells have been grown as organic triplicate cultures, and usually 2.5 million cells have been used per replicate per labeling experiment. Cells have been collected from tradition, washed of tradition medium and resuspended on ice-cold move cytometry buffer to which the Fc blocker was added for no less than 15 min. After blocking, cells have been adjusted to 1 million cells per ml of move cytometry buffer, after which the precomplexed antibodies have been added for staining at a remaining focus of two.5 µg ml−1. Staining occurred for 60 min at 4 °C on rotation, after which cells have been pelleted, supernatants have been discarded and cells have been washed as soon as with ice-cold 1× PBS. This wash is necessary to take away extra BSA within the move cytometry buffer. Subsequent, cells have been gently however rapidly resuspended in 980 µl of 100 µM biotin-phenol (Iris Biotech) in 1× PBS at 25 °C. To this, 10 µl of 100 mM H2O2 (Sigma-Aldrich) was rapidly added, tubes have been capped and inverted, and the response was allowed to proceed for two min at 25 °C. Exactly after 2 min, the samples have been quenched by including sodium azide and sodium ascorbate at a remaining focus of 5 mM and 10 mM, respectively. Samples have been inverted, and cells have been pelleted at 4 °C. Samples have been then washed sequentially as soon as with ice-cold move cytometry buffer after which twice with ice-cold 1× PBS, after which cell pellets have been lysed in 500 µl CLIP lysis buffer, briefly sonicated to solubilize chromatin and frozen at −80 °C for later processing.
As soon as all of the proximity labeling was full, lysates have been thawed in batches to carry out the next steps in parallel. Whole protein quantities have been quantified utilizing the BCA assay, and labeling effectivity and consistency have been checked utilizing western blotting. For biotin enrichment, we used the streptavidin western QC to find out the biotin sign throughout all samples first after which calculated the full µg of lysate that was wanted from that pattern to generate 5,000,000 models of streptavidin IR800 sign on the LI-COR system. This µg worth was then used because the enter mass for every of the replicates throughout all of the proximity labeling samples for biotin seize and MS preparation. Samples have been normalized to a remaining quantity of 500 µl with CLIP lysis buffer, and, to every pattern, 100 µl Pierce NeutrAvidin Agarose (Thermo Fisher Scientific) was added, and samples have been incubated at 4 °C for 4 h on rotation. Beads have been then washed twice with 1 ml of high-stringency buffer and twice with 1 ml of 4 M NaCl in 100 mM HEPES, all at 37 °C. Salts and detergents have been then rinsed from the beads by sequentially washing twice with 1 ml 1× PBS, twice with 1 ml of LC–MS-grade water (Fisher Scientific) and at last with 1 ml of fifty mM ammonium bicarbonate. An on-bead trypsin digestion was then arrange by including 200 µl of fifty mM ammonium bicarbonate and 1 µg MS-grade trypsin to every pattern and incubating for 16 h at 37 °C with occasional shaking. After digestion, the samples have been acidified by including formic acid at a remaining focus of 0.5%. The answer containing launched peptides was moved to a brand new tube, and the beads have been rinsed twice with 300 µl LC–MS-grade water to seize any remaining peptides. All elution and wash samples for a given replicate have been mixed and lowered to a quantity of <200 µl with a SpeedVac. Samples have been then desalted utilizing a C18 spin column (Thermo Fisher Scientific): preconditioned with 50% methanol in water and washed twice with 5% acetonitrile, 0.5% formic acid in water, the pattern was certain twice to the column, after which the columns have been washed 4 occasions with 0.5% formic acid in water. Lastly, peptides have been eluted into Protein LoBind tubes (Eppendorf) with two functions of 40 µl of 70% acetonitrile, 0.5% formic acid in water. Natural solvents have been eliminated with a SpeedVac, and samples have been absolutely dried with a lyophilizer. Ensuing peptides have been analyzed on the timsTOF Professional as described above.
For all peptides generated, we adopted the identical process for MS evaluation and peptide database looking. Particularly, a nanoElute was connected in line to a timsTOF Professional outfitted with a CaptiveSpray Supply (Bruker). Chromatography was performed at 40 °C by means of a 25-cm reversed-phase C18 column (PepSep) at a continuing move charge of 0.5 µl min−1. Cell part A was 98% water, 2% acetonitrile and 0.1% formic acid (vol/vol/vol), and part B was acetonitrile with 0.1% formic acid (vol/vol). Throughout a 108-min technique, peptides have been separated by a three-step linear gradient (5% to 30% B over 90 min, 30% to 35% B over 10 min, 35% to 95% B over 4 min), adopted by a 4-min isocratic flush at 95% for 4 min earlier than washing and a return to low natural situations. Experiments have been run as data-dependent acquisitions with ion mobility activated in PASEF mode. MS and MS/MS spectra have been collected with m/z 100 to 1,700, and ions with z = +1 have been excluded. Uncooked knowledge recordsdata have been searched utilizing PEAKS On-line Xpro 1.7 (Bioinformatics Options). The precursor mass error tolerance and fragment mass error tolerance have been set to twenty ppm and 0.03, respectively. The trypsin digest mode was set to semi-specific, and missed cleavages have been set to three. The human Swiss-Prot reviewed (canonical) database model 2020_05 (downloaded from UniProt) and the frequent Repository of Adventitious Proteins (cRAP model 1.0, downloaded from the International Proteome Machine Group) totaling 20,487 entries have been used. Carbamidomethylation was chosen as a set modification. Oxidation (M), deamidation (NQ) and acetylation (N terminus) have been chosen as variable modifications. Uncooked knowledge recordsdata and searched datasets can be found on the Mass Spectrometry Interactive Digital Setting (MassIVE), a full member of the ProteomeXchange Consortium, below the identifier MSV000092211. The entire searched datasets are additionally obtainable in Supplementary Info.
To establish enriched proteins from these datasets, we took an strategy that in contrast enrichment within the NPM1 to that of isotype antibodies. Outcomes of the database search have been first purged of non-human proteins and keratins as beforehand described. All proteins with fewer than two distinctive peptides have been additionally filtered out of the listing. Subsequent, Excel was used to calculate the imply spectral depend for every of the remaining proteins throughout triplicates. For every protein, the imply spectral counts related to every antibody probe have been divided by the imply spectral counts of the corresponding isotype, creating an enrichment issue of every protein within the proximity labeling over the isotype. Python (model 3.13) was then used to calculate the ratio of whole protein remoted from proximity labeling with protein-targeting antibodies divided by that collected with isotype antibodies. Proteins from every set of proximity labeling knowledge with enrichments both lower than two or the beforehand described ratio have been filtered from the dataset. The resultant lists comprise the hits related to every spherical of proximity labeling. To carry out GO time period evaluation, all proteins noticed throughout all 4 fractions from every cell line have been concatenated, creating an inventory of background proteins from every of the 4 cell traces examined. Lists of hits from the membrane hit pulldowns from every cell line have been submitted to DAVID ( and run in opposition to their respective backgrounds. Subsequent, for every class of GO time period (BP, CC and MF), the union of the highest 4 phrases throughout all cell traces and their related sizes and Benjamini values have been plotted for comparability of enrichment throughout cell traces.
Anti-NPM1 molecular counting
OCI-AML3 cells have been collected, washed with move cytometry buffer and resuspended at 106 cells per ml. They have been then blocked with Human TruStain FcX for 15 min with 5 µl of Fc block per million cells. They have been washed once more afterward. The cells have been then partitioned for dwell cell and glued cell staining. Cells deliberate for fixation have been stained with 1 µl of fixable violet LIVE/DEAD stain per 106 cells per ml for 30 min. Afterward, cells have been washed with move cytometry buffer and resuspended in 4% PFA for 10 min at room temperature. They have been washed with move cytometry buffer after which resuspended in 0.1% Triton X for five min at room temperature. Once more, the cells have been washed with move cytometry buffer. Dwell cells and glued cells have been then incubated with 500 ng FC8791–Alexa Fluor 647 in 100 µl for 30 min. Cells have been washed twice afterward, and dwell cells have been lastly resuspended in DAPI. For molecular counting, Quantum Alexa Fluor 647 molecules of equal soluble fluorochrome beads have been used from Bangs Labs. All samples have been analyzed on the BD LSRFortessa. Normal curves have been generated and fluorescence quantitation was carried out as per Bang Labs’ QuickCal evaluation device.
Cell floor proximity labeling of RNAs and gel evaluation
Samples have been ready right here in the same method as described above for the proximity labeling of proteins with key variations. The HRP-conjugated secondary antibody was exchanged for protein A–HRP (Cell Signaling) and used on the identical molar ratio: two main antibodies per one protein A–HRP molecule. The biotinylation reagent used right here was biotin-aniline (Iris Biotech); it was used at a remaining focus of 200 µM in 1× PBS for the labeling response, and the labeling response was allowed to proceed for 3 min at 25 °C. After pelleting the cells from the quenching response, cells have been immediately lysed, and whole RNA was remoted as described earlier than25. Briefly, RNAzol RT (Molecular Analysis Middle) was used to lyse cell pellets by putting the samples at 50 °C and shaking for five min. To part separate the RNA, 0.4× volumes of water was added, and samples have been vortexed, allowed to face for five min at 25 °C and lastly centrifuged at 12,000g at 4 °C for 15 min. The aqueous part was transferred to wash tubes, and 1.1× volumes of isopropanol was added. The RNA was then purified over a Zymo column (Zymo Analysis). For all column cleanups, we used the next protocol. First, 350 μl of pure water was added to every column, samples have been centrifuged at 10,000g for 30 s, and the flowthrough was discarded. Subsequent, precipitated RNA from the RNAzol RT extraction (or binding buffer precipitated RNA, under) was added to the columns, which have been centrifuged at 10,000g for 10–20 s, and the flowthrough was discarded. This step was repeated till all of the precipitated RNA was handed over the column as soon as. Subsequent, the column was washed 3 times in whole: as soon as utilizing 400 μl RNA Prep Buffer (3 M guanidine hydrochloride (GuHCl) in 80% ethanol) and twice with 400 μl 80% ethanol. The primary two centrifugation steps have been at 10,000g for 20 s, and the final one was for 30 s. RNA was then handled with proteinase Okay (Ambion) on the column. Proteinase Okay was diluted 1:19 in water and added on to the column matrix after which allowed to incubate on the column at 37 °C for 45 min. The column prime was sealed with both a cap or Parafilm to forestall evaporation. After the digestion, the columns have been dropped at room temperature for five min; decreasing the temperature is necessary earlier than continuing. Subsequent, eluted RNA was centrifuged out into contemporary tubes, and a second elution with water was carried out. The mucinase StcE (1.5 μg for each 50 μl of RNA, Sigma-Aldrich) was added to the eluate, and samples have been positioned at 37 °C for 30 min to digest. The RNA was then cleaned up once more utilizing a Zymo column. Right here, 2× RNA Binding Buffer (Zymo Analysis) was added, samples have been vortexed for 10 s, after which 2× (samples + buffer) of 100% ethanol was added, and samples have been vortexed for 10 s. The ultimate RNA was quantified utilizing a NanoDrop. In vitro RNase or sialidase digestions befell by digesting 50 μg whole RNA with one of many following: nothing, 4 μl RNase Cocktail (Thermo Fisher Scientific) or 4 μl of α2-3,6,8,9-neuraminidase A (NEB) in 1× NEB Glyco Buffer 1 (NEB) for 60 min at 37 °C. After digestion, RNA was purified utilizing a Zymo column as famous above and was then prepared for gel evaluation.
To visualise the labeled RNA, it was run on a denaturing agarose gel, transferred to a nitrocellulose membrane and stained with streptavidin25. After elution from the column as described above, the RNA was mixed with 12 μl of Gel Loading Buffer II (95% formamide (Thermo Fisher Scientific), 18 mM EDTA (Thermo Fisher Scientific), 0.025% SDS) with a remaining focus of 1× SYBR Gold (Thermo Fisher Scientific) and denatured at 55 °C for 10 min. You will need to not use Gel Loading Buffer II with dyes. Instantly after this incubation, the RNA was positioned on ice for no less than 2 min. The samples have been then loaded right into a 1% agarose, 0.75% formaldehyde, 1.5× MOPS Buffer (Lonza) denaturing gel. Exact and constant pouring of those gels is vital to make sure the same thickness of the gel for correct switch situations; we goal for solidified gels roughly 1 cm thick. RNA was electrophoresed in 1× MOPS at 115 V for between 34 and 45 min, relying on the size of the gel. Subsequently, the RNA was visualized with a UV gel imager, and extra gel was reduce away; leaving ~0.75 cm of gel across the outer edges of pattern lanes will enhance switch accuracy. The RNA was transferred with 3 M NaCl, pH 1 (with HCl) to a nitrocellulose membrane for 90 min at 25 °C. After switch, the membrane was rinsed with 1× PBS and dried on Whatman paper (GE Healthcare). Dried membranes have been rehydrated in Intercept Protein-Free Blocking Buffer, TBS (LI-COR Biosciences) for 30 min at 25 °C. After blocking, the membranes have been stained utilizing streptavidin IR800 (LI-COR Biosciences) diluted 1:5,000 in Intercept blocking buffer for 30 min at 25 °C. Extra streptavidin IR800 was washed from the membranes utilizing three washes with 0.1% Tween-20 (Sigma) in 1× PBS for 3 min every at 25 °C. The membranes have been then briefly rinsed with PBS to take away the Tween-20 earlier than scanning. Membranes have been scanned on a LI-COR Odyssey CLx scanner (LI-COR Biosciences).
Reporting abstract
Additional info on analysis design is on the market within the Nature Portfolio Reporting Abstract linked to this text.